Single and Pairwise Mutations and Their Impact on SARS-CoV-2 Proteins
Intro⌗
This past quarter (Spring 2020) has been an interesting one for sure. With the constant news of COVID and worldwide protests coming through every media outlet, focusing on schoolwork seemed like futile pursuit at times.
Thankfully, I was fortunate enough to have (mostly) fantastic professors that managed to provide informative and engaging lectures and assignments for the entirety of this challenging quarter.
A part of me is really enjoying working from home this quarter, and that part is mostly my GPA, which ended up being over an entire point higher than my unimpressive cumulative GPA. Not quite sure why or how that happened, but I guess I’ll just continue as I am…
The Class⌗
One of the things that made this quarter special is the fact that I was able to take my schools Bioinformatics class, which I have been trying to get into for a couple years.
It was taught by one of my favorite professors (and now my advisor), is only taught once a year, and requires a manual override to register for. I was thrilled when I found out I scraped one of the last slots!
This class was a unique opportunity to collaborate with other STEM students (bioilogy/chemistry) as well as grad students and tackle a real world research problem with techniques and tools we learn in the first part of the quarter.
The first 4 weeks brought all the CS people up to speed on the basics of DNA transcription/translation, protein structure, and alignment concepts.
The Project⌗
The latter half of the quarter was devoted to a collaborative research project, the groups for which we were allowed to self select out of a pool of pre approved project ideas.
The project I chose was this: Target various amino acids on the spike protein of the SARS-Cov-2, mutate them, and analyze the effectiveness of said mutations.
I chose this project because I like the idea of pinpointing vulnerable areas on the protein and mutating them to cause instability in the protein, possibly breaking functionality in the process.
My role in the project mainly consisted of managing communications between us and our professor, as well as creating the pipeline for our data. In short, the pipeline worked like this:
- Use
proMuteBatchto generate batch files forproMute, in order to exhaustively mutate multiple locations on the protein - Extract the output of
proMute(modified PDB files) as well as energy minimization data for each individual mutation (19 per target site) - Generate a script for the SDM tool, which allows us to predict $$\Delta\Delta G$$ values for each mutation.
I created a short bash script to automate this task. Although it could certainly be better optimized, I sacrificed some efficiency for readability by non linux nerds.
#!/bin/bash
# After making promute, copy this script into the build directory
# Change the "FILES" variable to the script (or scripts) you would like to execute
# to profile different mutation scripts, you can run this script with the "time" command
# Mutations on second PDB 6Y2E
./proMuteBatch 6Y2E A:A 25:25 X T25 # exhaustively mutate T at residue 25, chain A
./proMuteBatch 6Y2E A:A 26:26 X T26 # exhaustively mutate T at residue 26, chain A
./proMuteBatch 6Y2E A:A 27:27 X L27 # exhaustively mutate L at residue 27, chain A
./proMuteBatch 6Y2E A:A 28:28 X N28 # exhaustively mutate N at residue 28, chain A
./proMuteBatch 6Y2E A:A 29:29 X G29 # exhaustively mutate G at residue 29, chain A
./proMuteBatch 6Y2E A:A 30:30 X L30 # exhaustively mutate L at residue 30, chain A
./proMuteBatch 6Y2E A:A 31:31 X W31 # exhaustively mutate W at residue 31, chain A
./proMuteBatch 6Y2E A:A 32:32 X L32 # exhaustively mutate L at residue 32, chain A
declare -A residues
residues+=(
["PHE"]="F"
["LEU"]="L"
["ILE"]="I"
["MET"]="M"
["VAL"]="V"
["SER"]="S"
["PRO"]="P"
["THR"]="T"
["ALA"]="A"
["TYR"]="Y"
["HIS"]="H"
["GLN"]="Q"
["ASN"]="N"
["LYS"]="K"
["ASP"]="D"
["GLU"]="E"
["CYS"]="C"
["TRP"]="W"
["ARG"]="R"
["GLY"]="G"
)
sed -i 's/$/ em/' T25
sed -i 's/$/ em/' T26
sed -i 's/$/ em/' L27
sed -i 's/$/ em/' N28
sed -i 's/$/ em/' G29
sed -i 's/$/ em/' L30
sed -i 's/$/ em/' W31
sed -i 's/$/ em/' L32
# Customize with your scripts
FILES=("T25" "T26" "L27" "N28" "G29" "L30" "W31" "L32")
# Execute the lines in the script
for FILE in ${FILES[@]}; do
# Create a file to store SDM script
SDMFILE="${FILE}_sdm"
touch $SDMFILE
# Read each mutation into an array
readarray -t LINES < "$FILE"
# Routine to execute for every mutation
for LINE in "${LINES[@]}"; do
# Make a directory to store outputs
# Structure is PDBID.Chain.Location.Res2.output
OUTPUTDIRNAME=$(echo $LINE | awk '{ print $2"."$3"."$4"."$5".output" }')
echo [INFO] Creating Directory $OUTPUTDIRNAME
mkdir -p $OUTPUTDIRNAME
# Directory to look for EM files that were generated
# Structure is PDBID.ChainLocationRes2
EMDIRNAME=$(echo $LINE | awk '{ print $2"."$3$4$5 }')
# Actually call proMute
$LINE
# translate the promute command to an SDM script line
PDBID=$(echo $LINE | awk '{print $2}')
RESNUM=$(echo $LINE | awk '{print $4}')
CHAIN=$(echo $LINE | awk '{print $3}')
FINALRES=$(echo $LINE | awk '{print $5}')
ORIGRESCODE=$(cat ${PDBID}.pdb | grep ATOM | awk -v rn="$RESNUM" -v chn=$CHAIN '$6 == rn && $5 == chn {print $4}' | sed 's/.*\(...\)/\1/' | head -n 1)
ORIGRES=${residues[$ORIGRESCODE]}
if [ "$ORIGRES" != "$FINALRES" ]
then
# put this line in the script
echo $(echo $CHAIN $ORIGRES$RESNUM$FINALRES) >> $SDMFILE
fi
# Move all output files: PDB, Fasta, and EM data
mv *.txt $OUTPUTDIRNAME
mv *.pdb $OUTPUTDIRNAME
mv external/em/$EMDIRNAME $OUTPUTDIRNAME
mv $FILE $OUTPUTDIRNAME
echo [INFO] Moving files to $OUTPUTDIRNAME
done
# cleanup output
rm -rf "${FILE}_output"
mkdir -p "${FILE}_output"
echo [INFO] Cleaning outputs from last run
echo [INFO] Moving new files to "${FILE}_output"
mv $SDMFILE "${FILE}_output/"
mv *.output "${FILE}_output/"
done
Most of this script is pretty self explanatory. I simply generate a list of proMute commands using proMuteBatch, and executed all of them.
I used clues from filenames and commands (such as protein name and residue number) to generate unique directories of every mutation ran by proMute.
The only “confusing” part would be the generation of the SDM script. In short, the script format for SDM requires the original residue type for the location that is being mutated.
proMute does not care about or output this information, so I had to do some fancy pipelining to get it to work.
I look into the PDB file for the target protein, and find isolate all the ATOM lines, that correspond to individual atoms in the protein.
I then isolate the residue number of interest, making sure we are looking at the correct chain in the PDB file.
The first atom of the residue is kept, and then we simply look at the residue it belongs to. I wasn’t sure about the PDB notation, because sometimes one amino acid would be represented by multiple similar 3-4 letter codes.
So I just chopped off anything after three characters, and then run the three letter name through my dictionary, which spits out the single letter amino acid code required by SDM.
That’s a lot of work to go through for one letter…
The final step in the pipeline was a script created by an adjacent group studying double mutations in the same protein. This script analyzed the energy minimization data and used it to predict an effect on protein function.
Results⌗
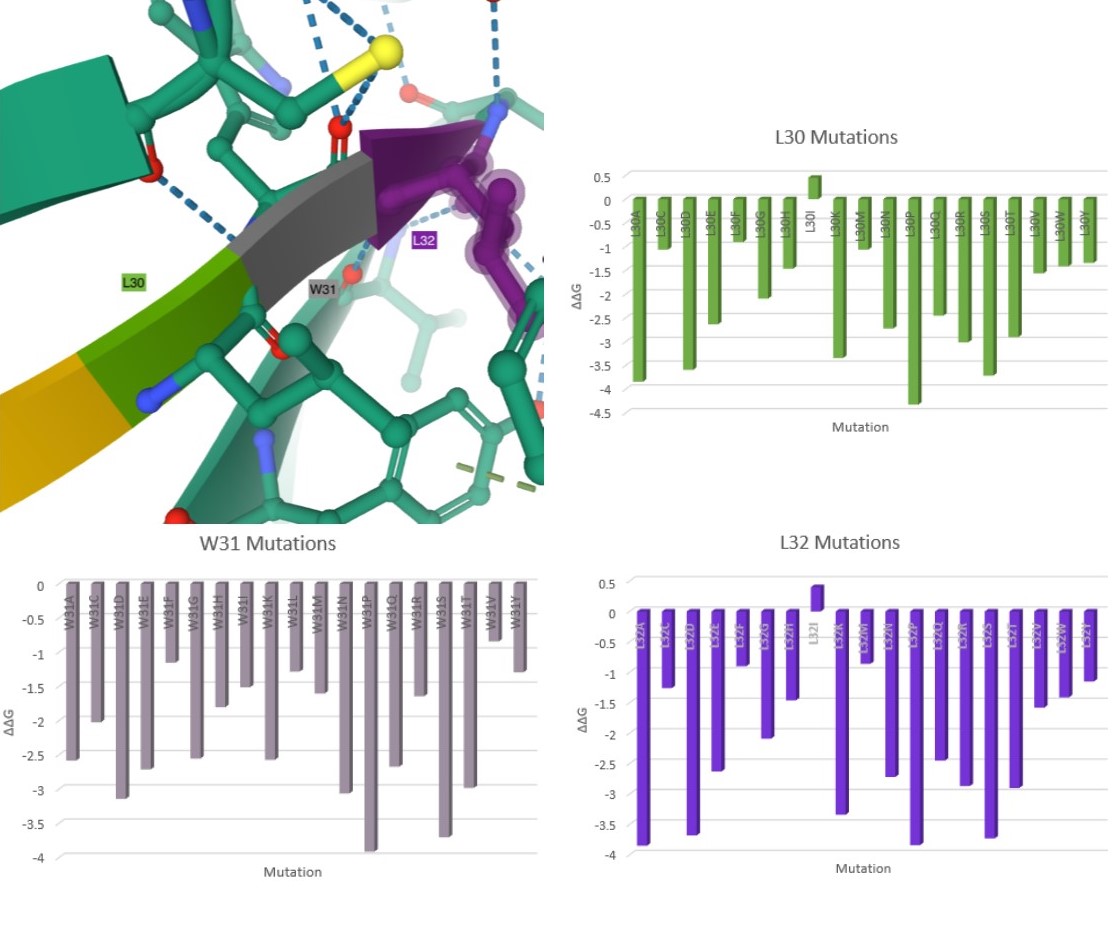
Here are my favorite figures from our final paper. We used pymol to color and label the locations of interest, as well as illustrate their bonds and connections.
The colored bar charts on the right represent one mutation for each of the 19 bars. The data plotted is the output $$\Delta\Delta G$$ produced by SDM, where a negative value indicates reduced stability in the protein.
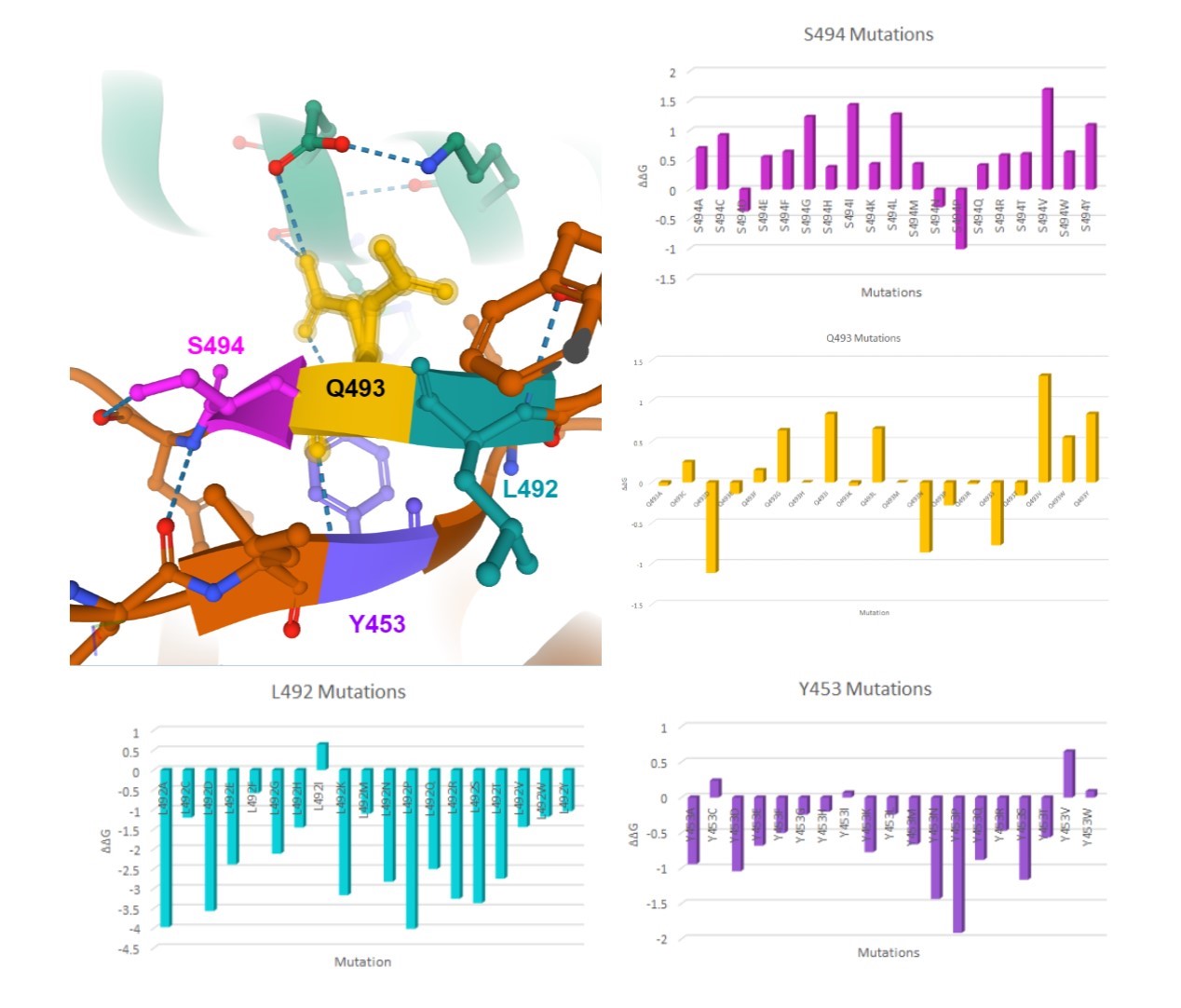

A heatmap visualization of the same data:
As you can see by the heatmaps, we were able to generate a significant amount of destabilizing mutations, especially on the 6Y2Estructure.
Conclusion⌗
If anyone wants to read our final project, I’ll leave it here to download. At the last minute, we decided to combine the reports of group 4 and 5, who investigated single and double mutations respectively. This provides a good way to compare a variety of possible mutations of the same structure.
I had a really great time in this class, and could not have dreamed of creating a project of this caliber, and I could not have done it without my awesome group members and my professor who had the perfect “hands off” approach where he would happily provide guidance, but left large decisions up to the groups themselves.